Pipelines
Pipelines are ordered chains of two or more SubSeq jobs. Use them for repeatable multi-step campaigns where each step can consume the previous step's output.
When To Use A Pipeline
- Use a pipeline for repeatable workflows such as design to sequence design to prediction to filtering.
- Use standalone jobs when you need manual inspection, branching, or a specific file choice between steps.
- Use batch-capable modes when a downstream step should consume a folder or output set from the previous step.
Example: Design-To-Review Pipeline
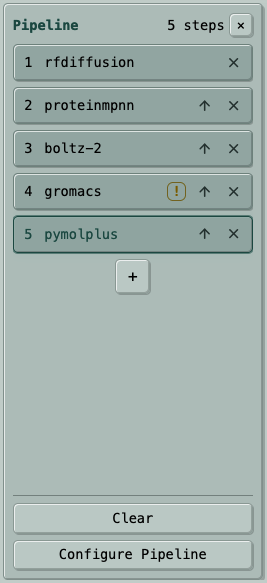
A practical protein-design pipeline can keep a candidate set moving through the same review logic every time:
- RF Diffusion generates candidate backbones.
- ProteinMPNN designs sequences for those backbones.
- Boltz-2 predicts and ranks the designed structures or complexes.
- GROMACS runs a relaxation, minimization, or short simulation check when that is part of the review plan.
- PyMOL+ renders static review figures from the final structures.
This kind of chain is most useful after you know the steps are compatible and you want repeatable campaign runs rather than one-off manual handoffs.
How Data Moves
- A pipeline is sequential: each step starts after the previous step finishes successfully.
- Step 1 can use the shared pipeline input when one is provided.
- Step 2 and later receive the previous successful step's main output as their input.
- If a step fails or is canceled, the pipeline stops with that terminal status.
Configuration
pipeline_input_srcisnone,upload, ordataset.pipeline_input_refis the dataset digest whenpipeline_input_srcisdataset.- Each step uses either structured
paramsor direct advancedargs. Do not mix both in one step. - Structured steps after the first should use a batch-capable mode when they consume a previous step's output folder.
- Per-step input and auxiliary source fields are not part of the public pipeline submit shape; use the top-level pipeline input.
Examples
- Design to predict to filter: generate backbones, design sequences, predict structures or complexes, then keep candidates above selected metric thresholds.
- Fold to figure: run a FASTA folder through a predictor, then render static review figures from the resulting structures.
- Batch processing: use a reusable dataset as the shared input, then chain a batch predictor, scoring step, and optional filtering step.
{
"project": "campaign-a",
"memo": "fold then render",
"pipeline_input_src": "dataset",
"pipeline_input_ref": "dataset_digest_here",
"steps": [
{
"program": "esmfold2",
"params": {
"mode": "fasta_inputs",
"fasta_inputs": "batch.fasta"
}
},
{
"program": "pymolplus",
"params": {
"mode": "batch_stills"
}
}
]
}Status And Outputs
- Track the pipeline status and each step's job ID from pipeline info.
- Final results usually come from the last successful job, but intermediate jobs remain useful for debugging and inspection.
- Use the normal job file, download, and manifest endpoints for per-step output files.
API Quick Reference
POST /api/v1/pipeline/submitsubmits a pipeline.GET /api/v1/pipeline/listlists pipelines.GET /api/v1/pipeline/infoinspects one pipeline and its step jobs.- Use job endpoints for files, logs, and downloads from each step.
Caveats
- Pipelines require at least two steps.
- Program compatibility matters: the next program must be able to consume the previous program's output format.
- Use standalone jobs if a human should choose one particular file between steps.